293
293
Concentration and Extinction Coefficient Determination for Oligonucleotides and Analogs Using a General Phosphate Analysis”,发表在1996年ANALYTICAL BIOCHEMISTRY杂志。由于翻译水平有限,详细内容请参考原文。
确定长度和序列(寡聚体)的核酸序列的合成和应用在过去十年中经历了显着的扩展。固相合成程序现在可以常规地提供具有天然存在的脱氧核糖或核糖核苷(A、G、C、和 T/U) 和主链连接(磷酸二酯),以及由这些部分的类似物组成的低聚物。在表征用于实验的低聚物样品时,通常需要获得浓度(c)和消光系数(ε)的准确值。对于具有常见碱基和核苷间连接的 DNA 和 RNA 寡聚体,目前有几种基于吸收的方法可以提供相当准确的ε和c测定。标准方法包括通过酶促或化学方法水解核苷间键,并将水解产物的吸收光谱视为游离残基吸收的总和。另一种方法需要将低聚物样品加热到高温(<80℃),以破坏任何二级结构或最近邻堆积相互作用,并使用与第一种方法相同的假设来估计残留物浓度。第三个分析考虑了在没有任何二级结构的情况下最近邻核苷残基之间的堆叠相互作用,并根据其序列估计低聚物的吸收率。这种方法,也许是最广泛使用的方法之一,由于多种原因在其对合成低聚物的一般应用中受到限制。这些包括将序列限制为仅具有标准核酸残基的序列、具有磷酸二酯以外的核苷间键的寡聚体的最近邻相互作用的变化,以及由于任何自互补性或二级结构而偏离预期的吸光度产率。
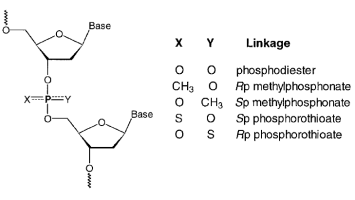
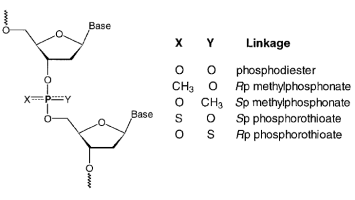
上面提到的第一个方法,我们称之为“核苷消解吸收”(Nucleoside Digest Absorption,简称NDA),是准确的。然而,为了广泛适用于具有不寻常主链或碱基组成的低聚物,NDA 要求核苷间键可以水解以产生游离残基,并且糖连接的杂环的吸收光谱在感兴趣的波长下具有良好的特征。核苷间连接的示意图如图 1 所示。根据取代基 X 和 Y,在磷的非桥接位置,形成三个共同的主链。磷酸二酯键是阴离子和非手性的。手性甲基膦酸酯和硫代磷酸酯键分别是非离子和阴离子的。对于 NDA 方法,通常使用酶(例如,蛇毒或脾脏磷酸二酯酶)来水解具有磷酸二酯主链的 DNA 和 RNA 低聚物。对于甲基膦酸酯低聚物,核苷间键对哌啶或氨水解敏感。硫代磷酸酯核苷间连接可被酶促水解。据我们所知,NDA 方法尚未用于确定具有硫代磷酸酯主链的低聚物的 c 或 e。大多数情况下,最近邻法已被用来估计这些数量。
材料和方法
化学试剂
所有化学品都是A.C.S.试剂级。碳酸钙、钼酸铵(VI)四水合物、抗坏血酸和2-氨基乙基膦酸来自Aldrich化学公司(Milwaukee, WI)。硫酸、高氯酸和硝酸购自Baxter Scientific Products(McGaw Park, IL)。A. P. H. A. 磷酸盐标准品[PO43-: 1.00 ml = 0.050 mg (50 ppm) P]由Ricca化学公司(Arlington, TX)提供。腺苷-5’-O-硫代单磷酸二锂盐由Calbiochem公司(San Diego, CA)提供。小牛胸腺DNA(钠盐)来自Sigma化学公司(圣路易斯,MO)。去离子蒸馏水(ddH2O)由Hydro Systems(Durham, NC)提供。
仪器
Labconco凯氏定氮消解仪,配有六个独立的恒温加热站(60011型,Fisher Scientific, Pittsburgh, PA)用于酸消解。可使用50或10毫升的凯氏管;清洗和重复使用这种玻璃器皿的结果重现性较差。一次性培养管(25 x 200 mm,Kimble 45060-25200,Fisher)可以替代凯氏管。用1 x 94英寸的玻璃纤维绝缘电热带(Thermolyne BIH101080,Fisher)包裹试管,有助于酸的进化。消解装置和加热带的温度用变阻器控制。仪器的冷凝器出口安装在一个冰冷的捕集器和一个吸水器上,使用耐酸管(PharMed AYX060-38和-22,Baxter)和一个氯丁橡胶塞子。从冷凝器出来的管子延伸到玻璃Erlenmeyer侧耳式捕集器中,并浸泡在氢氧化钠溶液中以收集和中和酸烟。放置在化学或通风橱内。带聚丙烯一次性吸头的可变容量吸管(Eppendorf, Brinkmann Instruments, Westbury, NY)用于等分标准溶液和未知溶液,并用于提起钼酸盐反应。聚苯乙烯血清学吸管用于制备和等分酸消解混合物以及制备试剂C。在瓦里安仪器公司(Sugarland, TX)的DMS100分光光度计上收集吸光度数据,分别使用1厘米的石英、塞子、掩膜杯(1毫升体积)和1厘米的玻璃杯(3毫升体积)进行uv和vis测量。
低聚物
磷酸二酯寡核苷酸是在Applied Biosystems(Foster City, CA)392 DNA合成仪上通过标准的磷酰胺化学方法合成的。一些甲基膦酸酯寡核苷酸是在同一系统上用甲基膦酸酯合成的。
在同一系统上用甲基膦酰胺化学法合成。硫代磷酸酯、混合骨架和三种甲基膦酸酯低聚物是Genta, Inc.(La Jolla, CA)的,以纯化形式获得。一般来说,磷酸二酯低聚物是在7M尿素、Tris-borate-EDTA缓冲液中通过制备变性PAGE进行纯化的。在某些情况下,使用Microsorb C18柱(Rainin Instruments, Woburn, MA)进行制备性反相HPLC,流动相为2-15%乙腈,50mM磷酸钠水溶液,pH 5.8。具有5'-磷酸二酯连接的甲基膦酸酯低聚物首先通过DEAE-纤维素色谱法从失败序列中分离出来,随后通过反相HPLC纯化。只有甲基膦酸酯连接的低聚物最初通过反相HPLC分馏并脱盐。最后通过正相HPLC在Cyclobond I 2000-β柱(Advanced Separation Technologies, Inc., Whippany, NJ)上进行纯化,使用由25mM乙酸铵水溶液和乙腈组成的等压缓冲系统。所有低聚物的离子交换和盐的去除是通过在50mM乙酸钠中吸附在Sep-Pak C18滤芯(Waters, Millipore Corp., Millford, MA)上,用15-30ml ddH2O洗涤,并用CH3CN: H2O(1: 1)洗脱到15ml聚丙烯管中来实现的。将低聚物溶液在离心蒸发器中浓缩,转移到1.5毫升的微离心管中,并进行干燥。将纯化、脱盐的低聚物溶解在ddH2O中,得到浓度为50到400μM的储备溶液。甲基膦酸盐低聚物溶解在CH3CN: H2O(1: 3)中,以帮助溶解和输送。所有的储备溶液都储存在0207C。低聚物的纯度通过分析HPLC验证,如果可能的话,通过放射性末端标记和变性PAGE进行验证。为了检查外来的磷酸盐,一些低聚物通过31P核磁共振光谱进行检测。
NDA方法
一式三份完整的低聚物样品(0.2 - 0.3吸光度单位)在1毫升标准缓冲液中制备;对于磷酸二酯类低聚物,10mM磷酸钠,10mM EDTA,pH7(缓冲液A);或者对于甲基膦酸酯类低聚物,以CH3CN:H2O(1:3)为溶剂的相同缓冲液(缓冲液B)。相应的三份样本在37℃下用0.2单位蛇毒磷酸二酯酶在50 μL的10mM Tris,2 mM MgCl2,pH8.2(磷酸二酯)中消解4小时,或用50ml的1M哌啶(甲基膦酸盐)消解。溶剂蒸发后,将消解物重新悬浮在1毫升的缓冲液A中。三个单独的酶或哌啶的样品被用作背景对照。在室温下记录所有样品的光谱,并获得任何更大吸收波长的吸光度值。根据朗伯比尔定律,消解后的低聚物Aλ,dig,的平均吸光度值用来计算其残留浓度cdig
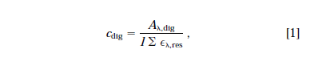
其中εel,res是低聚物序列中每个残基在所需波长下的消光系数之和。对于只含有DNA中典型碱基的寡聚体的分析,我们使用260 nm的波长和ε260,res(mM-1 · cm-1)的单核苷酸在低盐、pH7的条件下:dA 15.4;dC 7.4;dG = 11.5;dT = 8.7。对于含有不寻常的碱基的寡聚体,我们使用非典型核苷残基的最大吸收值和Beaven和同事报告的光谱中任何通常残基的内标法标定值。由于完整的低聚物的残留物浓度与消解物相当,因此它的ελ可以从其平均吸光度计算出来。消光系数的单位是M-1 · cm-1(l mol-1 · cm-1);但是,为了方便,我们以mM-1 · cm-1为单位报告,从而省略了103的系数。
GPA方法
试剂
大多数试剂在室温下是非常稳定的,只有抗坏血酸溶液必须储存在4℃,稳定期约为2个月。用ddH2O彻底冲洗干净的试剂瓶和烧瓶,以避免清洗剂对磷酸盐的污染。酸性消解混合物。50mg CaCO3(无水);90ml硝酸(69-71%);10ml高氯酸(69-72%)。6N硫酸:18毫升浓硫酸(95-98%);用ddH2O稀释到108毫升。2.5% 钼酸铵:2.5克(NH4)6Mo7O24·4H2O;溶于ddH2O;在容量瓶中带至100毫升。10%抗坏血酸:10g·L-抗坏血酸;溶于ddH2O;在容量瓶中加至100毫升。试剂C:2份ddH2O;1份6N硫酸;1份2.5%钼酸铵;1份10%抗坏血酸。计算开发当前样品所需的试剂总体积,并多加一些体积(样品数量×2毫升)。将各组分依次移入埃伦迈尔烧瓶中,每次加入后充分混合。需要现配现制。
酸性物质 消解过程
用于消解样品的试管在使用前用ddH2O冲洗并烘烤干。将所需的低聚物储备溶液的等分试样引入冷却的试管中--对于甲基膦酸盐低聚物,将吸头用一定体积的CH3CN:H2O(1:1)冲洗到试管中。将1毫升消解混合物分配到每个试管中。空白管只用酸混合液进行消解。将试管放在230℃下加热1小时。这是所有骨架转化为正磷酸盐所需的时间少。注意:在酸烟演化过程中,千万不要将冷凝器的任何位置打开。1小时后打开加热带,输出电压为120V。调整仪器设置,使其在1.5小时后获得≥300℃的温度。在2小时内,酸的混合物演化,在试管底部出现一个白色的白垩环。将试管放在185℃的干燥箱中,以驱除残留的酸,并防止引入水分。
磷钼酸盐比色法
在13支100毫米的硼硅玻璃试管中,用50ppm的标准磷酸盐溶液制备0.5至2.5μg·P的标准曲线(10至50μL)。在两支空白试管中加入2.0毫升ddH2O。在每个装有消解样品的冷却试管中也加入2.0毫升ddH2O。样品试管中再加入2.0毫升试剂C,摇匀使其溶解。在37℃下培养2小时,或在45℃下培养1.5小时。应避免更高的温度,以防止溶液蒸发损失。以样品和参比试管中标准曲线的两个空白点为零点,在820nm处记录所有样品的吸光度。
磷酸盐浓度的测定
所有数据均使用KalideaGraph 3.0软件进行分析。如果有问题的点的偏差是标准偏差的三倍,那么线性回归曲线中的离散点将在95%的置信度下被丢弃。有两种方法可以将未知溶液的数据与标准曲线进行比较,以获得该溶液的浓度。
(i) 斜率的比例。待分析的低聚物溶液以递增的体积分布在五个试管中,估计范围不超过2.5μg的磷,并消解掉一个没有低聚物的试管作为空白。比色分析后,用消解后的低聚物的吸光度减去空白吸光度。得到线性回归(A820 vs ml),未知低聚物溶液的磷含量ΓP(μg/μl),计算如下:
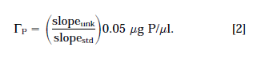
(ii) 内标法。一式三份的待分析的低聚物溶液的等分试样与三个空白试样一起消解。使A820值在0.2至0.5单位之间(1至2.5μg· P)。编制标准曲线,确定斜率(A820 vs μg P)。消解后的低聚物溶液的平均读数减去平均空白值,得到A820,unk。未知溶液中磷的总微克数从标准曲线中内标值为A820,unk(slopestd)-1,ΓP通过将此数量除以分析中消解的体积(ml)得到。
低聚物的浓度和消光系数
在获得低聚物溶液的GP值后,低聚物的摩尔浓度可计算为
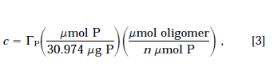
其中n是低聚物中磷原子的化学计量数。大多数情况下,n会比序列中的残基数量少一个。用完整的寡聚体的Aλ, 根据上述NDA方法计算ελ,同时考虑到制备溶液所需的稀释因子。
结果和讨论
总磷酸盐的回收率
GPA 根据序列中的化学计量磷确定低聚物的浓度,使总磷酸盐的回收成为该方法最重要的特征。 为了确认引入消解管中的磷在湿灰化后以正磷酸盐的形式定量存在,整个方案是对磷酸盐标准本身进行的。 在 5 个不同的日子里,从标准溶液中制备了两组磷酸盐样品 (0–2.5 mg)。 一组按常规程序消解,另一组用作标准曲线。 消解曲线和标准曲线的回归常数列于表 1。还给出了每次试验曲线斜率的百分比差异,该值表示通过该方法检测的处理和未处理样品的磷酸盐量之间的一致性。在每种情况下,消解曲线和标准曲线都给出相似的斜率,任何试验的很大差异约为 2%。平均差异几乎为零,标准偏差为 1.5%。经处理和未经处理的样品之间没有系统差异。标准曲线比消解曲线具有更好的相关性,考虑到对后者的额外操作,这是一个合理的观察结果。该分析表明,开发的协议能够以准确和精确的方式定量说明添加到消解管中的所有磷酸盐。
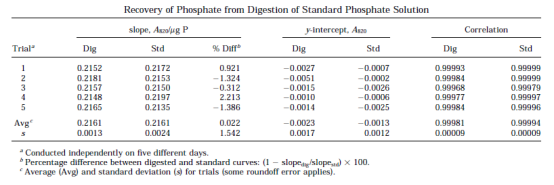
表1 标准磷酸盐溶液消解后的磷酸盐回收率
模型化合物的分析
在感兴趣的低聚物中模拟磷酸盐连接的化合物被选为模型,以评估磷酸盐衍生物转化为正磷酸盐的情况。小牛胸腺DNA是具有磷酸二酯骨架的寡聚体的模型,需要水解3'-和5'-酯键到糖。为具有甲基膦酸盐骨架的低聚物选择的模型是2-氨基乙基膦酸(AEP)。这个分子被Kirkpatrick和Bishop用来研究有机磷酸盐向正磷酸盐的转化,他们发现需要高氯酸来实现磷碳键的氧化。腺苷-5'-硫代单磷酸酯(AMP-S)作为具有硫代磷酸酯骨架的低聚物的模型,需要氧化磷-硫键。制备了≈0.5mg/ml的小牛胸腺DNA溶液,并根据报告的每个残基ε257的6.6 mM-1 · cm-1确定其浓度。AEP和AMP-S的水溶液是在50ppm磷的条件下通过重量法制备的。三个模型化合物的分析结果见图2。测试溶液中的磷含量与预测的曲线相当吻合。对未消解的样品进行比色分析时,没有吸光度。这些结果表明,所开发的方法应能将含磷酸二酯、膦酸酯和硫代磷酸酯的低聚物中的磷定量地转化为正磷酸盐。
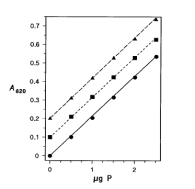
图2. 通过磷钼酸盐复合物的形成对模型化合物的酸消解物进行比色分析。小牛胸腺DNA,观察(●)和预测(-)。AEP,观察(■)和预测(- - -)。AMPS,观察(▲)和预测(- -)。AEP和AMP-S的数据分别偏移了0.1和0.2吸光度单位。
低聚物的一般磷酸盐分析
由典型核苷组成的序列。对具有四种不同类型骨架(磷酸二酯、甲基膦酸酯、硫代磷酸酯和混合)的低聚物进行了ε260的评估。表2给出了用GPA和NDA得到的结果,以及用Cantor等人和Puglisi和Tinoco的最近邻近似法计算的序列。对于一些低聚物,通过GPA对原液中磷含量的多次测量给出了4%或更少的标准偏差,证明了该方法的可重复性。一般来说,由GPA和NDA确定的ε值都很一致。最有趣的比较是由实验方法确定的经验值和基于预测方法的计算值之间的比较。对于磷酸二酯低聚物--除了同源嘧啶序列(No.1)--所有序列的测量的ε260值都明显低于预测值。一个合理的解释是,这些低聚物有额外的堆积相互作用,导致260nm处的吸光度降低。例如,交替的d-(AG)二核苷酸序列(编号2)的实验测量系数比预测值低20%左右。众所周知,含有这种图案重复的序列在各种环境条件下都具有自结构。相比之下,甲基膦酸酯低聚物测得的ε260值通常与预测值接近--其中两个案例(11号和12号)的值有点高。具有非离子骨架的低聚物的这种一致程度可能是几个因素的偶然结果。核苷连接处没有电荷可能会加强单链状态下的堆积效应,这应该会导致系数降低。然而,用于测量甲基膦酸盐吸光度的溶剂含有乙腈,它可能作为一种药剂破坏堆积作用。此外,非离子低聚物的磷上的外消旋甲基可能会影响与相应的磷酸二酯序列中发现的内在结构差异。作为一个群体,具有硫代磷酸酯和混合骨架的低聚物的ε260值低于预测值,这也可能是由于单链状态下的结构比具有天然骨架的序列的预测值要高。混合骨架的低聚物(No.17)是一个有趣的分子,因为在19个核苷内连接中,6个是磷酸二酯,7个是甲基膦酸酯,6个是硫代磷酸酯。此外,糖分子既是2'-脱氧核糖又是2'-O-甲基核糖。用NDA分析水解所有的连接物需要一系列具有挑战性的酶和化学反应。通过一个简单的消解和比色程序,GPA可以为这个复杂的序列提供c和ε的定量。
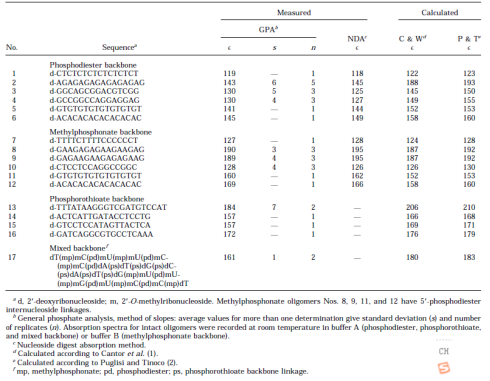
表2 用不同方法测定的低聚物消光系数(ε260, mM-1 · cm-1)。
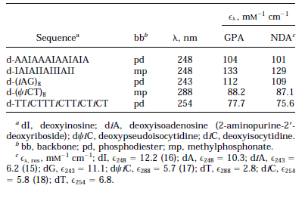
含有非典型核苷的序列。图3A显示的是脱氧腺苷(dA)核苷及其类似物脱氧异腺苷(diA,也被称为2-氨基嘌呤核苷)的紫外光谱。这些光谱说明了NDA可能不适合于确定浓度和消光系数的典型情况。类似物diA的最小吸光度在260nm附近,正好是标准残留物dA的最大值。此外,除了diA的λmax(220nm),模拟核苷在通常的碱基具有明显更大的ε的波长范围内具有相对低的吸光度。当diA作为二分之一的残基被纳入低聚物d-(iAG)8时,其光谱与观察到的母序列d-(AG)8的光谱有相当大的变化(图3B)。表3列出了五个具有磷酸二酯或甲基膦酸酯骨架的低聚物的消光系数,这些低聚物含有非典型的残基。除了上面讨论的d-(iAG)8低聚物外,还有两个含有脱氧肌苷(dI),一种天然存在的小核苷;一个含有脱氧假异胞苷(dφiC),一种合成的C核苷;一个含有脱氧异胞苷(diC),一种合成的嘧啶类似物.两种方法得到的消光系数对所有被检测的低聚物都很一致。
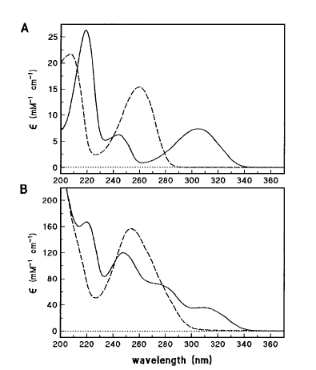
图3. 室温下缓冲液A中的吸光光谱。
(A) 核苷2'-脱氧异腺苷(-)和2'-脱氧腺苷(--)。(B) 磷酸二酯骨架低聚物d-(iAG)8(-)和d-(AG)8(--)。
内标法与斜率的比率。表2和表3中报告的所有GPA值都是通过斜率比得到的。我们更喜欢这种方法,因为它提供了未知溶液的样品量范围内的数据。另一方面,内标法也是广为人知的,而且容易检测。为了比较这两种方法,我们通过内标法测量了以下五种低聚物的εS值(mM-1 ·cm-1)。(i) d-GGCAGCGGACGTCGG(表2,第3行),ε260=131;(ii) d-(AC)8磷酸二酯(表2,第6行),ε260=144;(iii) d-GAAGAGAGAAGAAGAG(表2,第8行),ε260=188。8),ε260=188;(iv)d-(GT)8甲基膦酸盐(表2,第11行),ε260=159;(v)d-AAIAAAAIAIA(表3),ε248=103。与通过斜率比得到的数值比较表明,这两种方法得到的结果相似。
表3 含有非典型残基的低聚物的消光系数
其他溶质的干扰
除了含磷的样品外,其他样品成分的存在长期以来一直是生物组织样品中磷酸盐比色法的一个问题。我们的储备溶液可能只包含溶剂、溶解的低聚物和任何相关的反离子。有时可能需要使用GPA对含有缓冲剂和/或其他溶质的溶液中的低聚物进行定量。然而,这些物质的存在可能会干扰分析,因为它们会阻止湿灰化程序消解低聚物或干扰比色法。为了解决这个问题,我们检查了10种不同的溶液,这些溶液是在实验室中随机收集的,已经准备好用于光谱、电泳、色谱和杂交实验。酸性消解和比色分析是在0. 1毫升的溶液,其中含有以下溶质的不同组合[最大浓度]:醋酸根离子[50 mM];硼酸根离子[90 mM];氯离子[150 mM];柠檬酸根离子[15 mM];EDTA[10 mM];氢氧化钠[50 mM];镁离子[20 mM];Mops[50 mM];Pipes[50 mM];钾离子[140 mM];亚精胺[0. 2 mM];精胺[1 mM];钠离子[150 mM];和Tris[90 mM]。含有额外成分的消解液的平均吸光度与单独用0.1ml ddH2O制备的空白液相同。使用相同的溶液和空白板,随后对来自磷酸盐标准、AEP和AMP-S溶液的2.5 μg磷进行消解。消解后,对磷酸盐标准品检测到了预期的磷酸盐量;然而,在AEP和AMP-S样品与含有大量有机物的缓冲液混合的情况下,吸光度低了几个百分点。在这些消解过程中,注意到试管中出现了发烟的硝酸(黄白色至棕色的烟)。随后用1.5毫升的酸消解混合物重复灰化过程,并通过比色法计算出预测的磷含量。在这些溶液存在的情况下,对消解的低聚物进行进一步的试验,当消解混合物的量因此增加以说明额外的氧化负荷时,得到了预期的数值。消解甲基膦酸盐溶液会产生来自乙腈的额外残留物;但是,比色分析不受影响。这里开发的方法可以成功地应用于含有许多缓冲液中的各种额外成分的低聚物溶液。如果含有其他溶质的低聚物样品要用GPA进行检测,建议对溶液本身进行背景分析。显然,含有磷酸根离子(或其他磷衍生物种)的溶液是不兼容的。含有金属氧化物的缓冲液(如可可碱)会在钼酸盐反应中产生颜色,应避免使用。
误差的来源
实验者关心的是磷既不丢失也不增加。消解过程中磷酸盐的损失可以通过在酸混合物中加入钙离子来防止。用清洁剂清洗过的玻璃器皿可能会造成额外引入的磷酸盐,使用原始的消解管、清洗干净的试剂玻璃器皿和一次性吸管可以避免。另一个来源可能是外来磷酸盐引入的。在我们最初的许多分析中,在低聚物储备溶液中检测到的磷酸盐含量比紫外吸收率所能检测到的的要多,导致GPA获得的ε明显减少。我们通过31P核磁共振检查了一些储备溶液,以调查含磷物种的类型。在GPA的磷含量异常高的每种情况下,都观察到无机磷酸盐核磁共振信号。所有这些储备溶液都是在一个多次使用的C18柱上脱盐的,该柱子反复暴露在大量的磷酸盐缓冲液中。怀疑这个纯化步骤是外来磷酸盐的来源,我们随后在一次性使用的C18柱上单独脱盐了所有低聚物,使用醋酸钠缓冲液作为水离子相来吸附样品。这种处理完全解决了磷酸盐污染的问题,因为核磁共振只检测到参与核苷间连接的磷酸盐物种。随后的GPA导致ε值与NDA提供的值有更好的一致性。
这里考虑的所有方法(GPA、NDA和最近邻计算)都依赖于对低聚物序列的了解;因此,样品的纯度和均匀性很重要。此外,任何低聚物的吸收光谱可以被大量的环境条件随意调节,其中包括浓度、温度、pH值、缓冲液、离子强度、有机溶剂和特定离子。因此,在检测消光系数时,必须说明测量吸收光谱的条件。分析测试中,我们在室温下使用低离子强度、中性、磷酸盐缓冲溶液,并加入少量EDTA以螯合二价阳离子。这些条件通常与带电的骨架的低二级结构兼容,能得到很好的重现性。对于非离子型甲基膦酸酯低聚物,当只用水作为溶剂制备储备液和缓冲液时,光谱数据的重现性很难做到很好。当在这些溶液中加入25%(体积)的乙腈作为助溶剂时,吸光度数据的重现性显著提升。
评论
对于NDA和GPA,必须消解一部分低聚物。可以估计每种方法所需的数量。从表2来看,平均残留物ε260为9 mM-1 · cm-1,这意味着NDA分析至少需要消耗0.07 μmol的残留物。正如这里所开发的,GPA可以在低至0.1μmol P(约等于残留物)上进行。因此,就低聚物成本而言,这两种分析方法大约是等价的。对核酸样品进行浓度和消光系数定量的磷分析,在文献中已有几十年的历史。然而,这种方法目前在相对较少的实验室中被使用。最近,又有了另一种分析方法,采用酶介导的正磷酸盐和鸟苷类似物的转化来产生不同的吸收物种。一种不需要预消解的磷定量方法是ICP(电感耦合等离子体)分析。这两种方法的检测限都与磷钼酸盐法提供的范围相当。考虑到材料的可用性和仪器的成本,可以认为GPA在低聚物上有实际的应用。
GPA可以消解和化验二磷和三磷核苷酸中含有的磷。根据腺苷的吸光度,ADP和ATP的分析结果分别为两个和三个正磷酸盐的当量。灰化过程也很容易将亚磷酸酯分子[P(III)氧化态]氧化成正磷酸盐。胸苷亚磷酸酯和甲基亚磷酸酯(用于固相低聚物合成的化合物)的溶液通过GPA进行检测。根据三酰反应为这些溶液确定的浓度,亚磷酸酯被定量地转化为正磷酸盐。因此,如果样品中含有此类形式的磷,则应在分析中加以考虑。
结论
这里开发的一般磷酸盐分析法提出了一个便宜的、标准的消解和比色方法,对任何生物化学或分子生物学实验室都是实用的。用这种方法,具有磷酸二酯、甲基膦酸酯、硫代磷酸酯或这些骨架组合的低聚物溶液可以用磷和低聚物浓度来表征。如果已知该低聚物在规定条件下的吸收光谱,那么就可以得到任何波长的消光系数。这种方法不需要事先了解给定序列中各个核苷残基的吸收谱,因此,与其他获得核酸及其类似物的浓度和消光系数的定量和预测方法相比,具有优势。目前丰富的低聚物化学成分包括许多基于磷的骨架类似物。这里开发的GPA很可能也适用于这些分子中的许多分子。
网 址:www.bioprocessanalytics.com
邮 箱:info@bioprocessanalytics.com



长按屏幕识别二维码
打开手机扫描二维码