392
392
摘要
本研究的目的是把Edelhoch方法测得的蛋白药物消光系数(EC)的实验值与蛋白药物消光系数的理论值进行比较。文章对超过176种观察结果进行了广泛的分析,涵盖了19种不同结构类型的分子,包括单克隆抗体、双特异性抗体、融合蛋白和BiTE。通过评估每种类型分子测量的重复性和相对标准偏差(%RSD)来确定测量的精度。结果显示Edelhoch方法的RSD为1.7%,平均RSD为0.9%。对于实验值与理论值的偏差,即平均实验消光系数与理论消光系数的差值,观察到的更大偏差为5.3%,平均偏差为2.6%。文章研究结果表明,与常用的氨基酸分析(AAA)方法相比,Edelhoch方法可靠性高,显著提高了效率,降低了时间和成本,并提高了安全性。
介绍
对于大多数生化、生物物理和生物实验来说,准确测定蛋白质浓度是非常必要的。测定蛋白质浓度最常用的方法是测定280 nm处的吸光度,根据280 nm处的分子特有的消光系数,并应用Beer-Lambert定律进行测定[1]。只要消光系数(ε280)是准确的,这种测定蛋白质浓度的方法就是有效的。而精确测定消光系数则又需要精确测量蛋白质浓度。测定蛋白质浓度的常用方法有:(a)氨基酸分析法[2],(b)定氮法[3],(c)干重法[4]和(d) Edelhoch法[5]。(目前,有一种革新的光谱技术SoloVPE斜率光谱法,被用于Edelhoch方法中的浓度检测,SoloVPE也可直接进行消光系数的检测)
Edelhoch方法是基于比较两组样品的吸光度,一组在折叠状态下,另一组在相同蛋白浓度的展开条件下[5-7]。由于折叠和未折叠的蛋白质的浓度相同,因此Beer-Lambert定律可以表示为:
Af/Au = εf/εu
其中,Au是未折叠蛋白的测量吸光度,Af是已折叠蛋白的测量吸光度,εf为折叠蛋白的实验消光系数,εu为用等式(4)确定的未折叠蛋白的消光系数,该研究是基于对6M盐酸胍中色氨酸、酪氨酸和半胱氨酸的合适模型化合物的Edelhoch实验研究[5-7]。因此,根据Edelhoch方程可确定折叠蛋白的实验消光系数:
εf = εu*(Af/Au)
对于由标准氨基酸组成的典型蛋白质,除了来自二硫键的贡献外,在280 nm附近波长处的吸收归因于三个氨基酸生色团:色氨酸、酪氨酸和苯丙氨酸[8]。对于没有色氨酸或酪氨酸残基的蛋白质,Scopes方法[9]可以通过在205 nm处的吸光度来测量蛋白质浓度。
Pace等人在1995年进行了详细的研究,并比较了不同的实验方法来确定蛋白质的消光系数[10]。他们的研究表明,Edelhoch方法由于其高准确性、高效率和高重现性,是测量蛋白质实验消光系数的可靠方法。在同一研究中,Pace等人还开发了一个确定蛋白质水溶液消光系数的高精度方程,该方程目前被广泛用于确定蛋白质的理论消光系数或预测消光系数:
ε280 = 5500 × (# of Trp residues) + 1490 × (# of Tyr residues) + 125 × (# of S-S bonds)
Pace等人在1995年发表的研究结果,是基于使用不同的正交实验方法,对大量实验测出的消光系数进行统计分析。该研究使用的数据涵盖了80种不同的球状蛋白的116个测量消光系数,但该研究未纳入抗体数据或蛋白药物数据。后来,在2015年,Maity等人[11]将Edelhoch方法应用于单克隆抗体,实验表明该方法对这类分子表现良好,同时表明Pace方程也适用于单克隆抗体。在我们的研究中,进一步扩展了Edelhoch方法的应用,不仅包括单克隆抗体(mAbs),还包括其他结构类型的蛋白药物,如双特异性抗体,BiTE分子(双特异性T细胞抗体)和融合蛋白来测试Edelhoch方法的性能。单克隆抗体是进入临床研究的且增长最快的治疗方法之一[12]。目前,生物制药领域正在经历从传统的单克隆抗体到基于IgG折叠的多种模式的转变,如BiTE、融合蛋白和双特异性抗体,这有助于治疗模式的多样性[13]。传统的单抗是一种Y型分子,携带两个可变的Fab结构域,可以结合到目标抗原和免疫刺激Fc区域,帮助激活免疫反应。虽然标准单克隆抗体有两个臂结合同一目标抗原,但另一方面,双特异性抗体被设计成两个臂可以结合两种不同的抗原[14]。融合蛋白通常由一个IgG Fc结构域与受体(靶区)的结合域融合组成[15]。Fc区域提供更长的半衰期,靶区域决定其治疗效果。BiTE是生物药物治疗的新方法之一。它们是通过连接来自Fab区域的轻链和重链的一对可变域的目标区域而创建的[16,17]。一只手臂可以与细胞毒性T细胞上发现的蛋白质结合,另一只手臂可以与肿瘤细胞上的特定蛋白质结合,从而实现肿瘤细胞与T细胞的连接。鉴于生物药物治疗模式的日益多样性,使用不同结构类型的分子来测试生物表征方法的有效性是很重要的。我们研究了对于不同结构类别的蛋白药物,使用Pace方程预测的理论消光系数,与用Edelhoch方法预测的实验消光系数进行比较。除了提供不同蛋白药物的Edelhoch数据外,我们还将Edelhoch数据与常用的氨基酸分析(AAA)方法数据进行了比较,并对这两种方法的精度、复杂性,以及效率和安全性进行了总体展望。
材料与方法
材料
所有纯化蛋白均来自Amgen公司,Dulbecco的磷酸盐缓冲液(PBS),自制6.6M盐酸胍(来自Sigma-Aldrich)的PBS,盐酸和苯酚来自ThermoFisher公司,β-巯基乙醇来自Gibco,非亮氨酸来自Sigma-Aldrich。
方法
理论摩尔消光系数由氨基酸组成计算得出[10],公式为:
ε280 = 5500 × (# of Trp residues) + 1490 × (# of Tyr residues) + 125× (# of S − S bonds)
1 mg/ml蛋白质的理论消光系数是通过除以摩尔消光系数的分子量来确定的。Edelhoch方法是由Grimsley GR等人在《Current Protocols in Protein Science》(2003)[7]中描述的。简单地说,用PBS(pH7.2)透析蛋白质原液过夜。透析后的浓度使用SoloVPE(斜率光谱技术)进行测量,然后将透析蛋白的原液适当稀释成磷酸盐PBS缓冲液(用于折叠蛋白吸光度测定)和6.6M盐酸胍PBS缓冲液(用于未展开蛋白吸光度测定),保持最终稀释的浓度相同。这些稀释可以通过手动移液或使用TECAN机器人系统进行移液。将稀释后的样品轻轻混合4-5次,在室温下静置30 min,然后用Cary60紫外可见分光光度计在200-400 nm的光谱范围内测定折叠和展开样品的吸光度。根据《Current Protocols in Protein Science》(2003)所述[7],还需测定缓冲液的吸光度作为空白,并对蛋白样品进行散射光校正。稀释并使最终测量的Abs280在0.1-1个单位的范围内,即在Beer-Lambert定律的线性区域内。使用以下方程(方程(4)),根据色氨酸、酪氨酸和二硫键的数量计算出6M胍中未展开蛋白的消光系数。该方程(4)的原理利用的是6M胍盐酸中这些残基的消光值,然后由Edelhoch方法根据合适的化合物模型得出的[5–7]。
ε280 = 5690 × (# of Trp residues) + 1280 × (# of Tyr residues) + 120 × (# of S–S bonds)
标准偏差计算为:
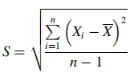
其中,S=样本标准差,n=观察次数,Xi=分子的观测值,`x=观察的平均值
相对标准差(RSD)用标准差除以平均值计算,用%表示。标准差偏(SE)的计算方法是将标准偏差除以观测次数的平方根。
AAA按照Crabb JW等人[18]所述进行。简单地说,用PBS透析蛋白质原液,取约50 μg的样品放在玻璃瓶中,使用快速真空浓缩器干燥。随后,在没有空气的情况下(充氮气),样品在控制条件下(6N盐酸,5%苯酚和β-巯基乙醇)仔细地进行水解。然后将水解管置于110 oC的烘箱中放置24小时,然后放在冰上放置10 min,再次使用快速真空浓缩器干燥。然后将样品在特定体积的正亮氨酸缓冲液(0.1 mM正亮氨酸溶解在0.2N盐酸中)中小心重悬、过滤,然后载入氨基酸分析仪(日立L8900)。报告结果使用的是氨基酸报告程序(EZ Chrom Elite™)。
结果与讨论
Edelhoch方法
分析数据包括跨多个分子类型(单分子抗体、双特异性抗体、融合蛋白、BiTE)的176个观察结果,如表1所示。本研究中使用的单抗分子的分子量为144 kDa至147 kDa,色氨酸残基数量为22-26,酪氨酸残基数量为50-66,二硫健数量为16-18。双特异性抗体的分子量为159 kDa~196 kDa,色氨酸残基为27~36个,酪氨酸残基为58~82个,二硫健为16~20个。融合蛋白的分子量为82 kDa~122 kDa,色氨酸残基为10~20个,酪氨酸残基为24~40个,二硫健为10~24个。BiTE的分子量为104 kDa~132 kDa,色氨酸残基为20~24个,酪氨酸残基为45~56个,二硫健为12~16个。对每种分子类型进行多次测量,至少重复3次或更多,以监测不同时间、仪器和分析人员之间的变化。对于每种分子类型,使用Pace等人提出的公式(3)计算理论消光系数,并使用Edelhoch方法确定观察到的实验消光系数。通过评估每种分子类型的可重复性来测量精度,并通过计算标准偏差(SD)、相对标准偏差(RSD)和标准误差(SE)来进行确认(表1)。理论消光系数的偏差是通过计算平均实验消光系数与理论消光系数之间的差值来确定的(表2)。
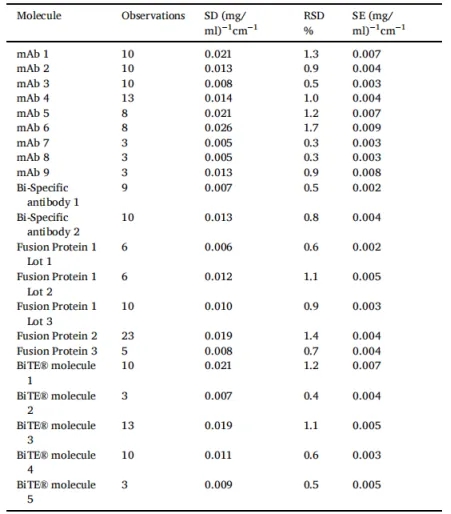
表1 用Edelhoch方法进行实验消光系数的检测,包括SD、RSD%和SE值。
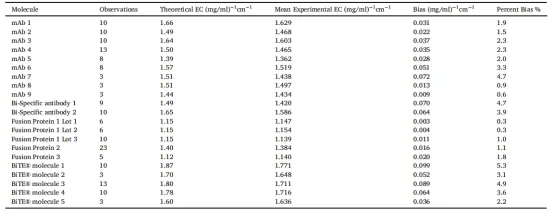
表2 用Edelhoch法计算的实验消光系数与理论消光系数的比较。
从RSD值(表1)和百分比偏差值(表2)可知,Edelhoch方法具有较高的精度/重复性,与理论消光系数的偏差较小。在我们的研究中,观察到的最高和最低的RSD分别为1.7%和0.3%,平均RSD为0.9%。对任何给定分子的百分比偏差表示的理论消光系数的高偏差和低偏差分别为5.3%和0.3%,平均偏差为2.6%。总的来说,我们的数据与Pace等人在1995年[10]和Maity等人在2015年[11]上发表的研究结果非常一致。因此,这些研究表明,Edelhoch方法是确定不同结构类蛋白药物的可靠方法,包括单克隆抗体、融合蛋白、双特异性抗体和BiTE。但是,需要记住的是,如果目标蛋白质不含色氨酸或酪氨酸,则不能应用Edelhoch方法来确定蛋白质的消光系数。在这种情况下,消光系数可以通过应用Scopes法[9]测量在205 nm处的吸光度来确定消光系数。
Edelhoch方法与AAA方法的比较
氨基酸分析法(AAA)是测定蛋白质[18,19]实验消光系数的常用技术。在该方法中,蛋白质或肽被恒沸的盐酸水解消化,得到的氨基酸通过强阳离子交换色谱和柱后印三酮衍生分析。然而,尽管在原则上很简单,但在水解步骤和随后的色谱和衍生化步骤[20,21]中使用AAA方法存在几个挑战。例如,水解后除水不充分以及水解过程中可能的氧化性破坏,此外,一些氨基酸(主要是芳香族氨基酸)会被破坏。综上所述,AAA方法是复杂的,高度依赖于分析人员的处理水平,需要分析人员长时间高度警惕,并且在水解步骤中会使用到危险物质(6N盐酸、β-巯基乙醇、苯酚等)[19] 。因此,在开始使用AAA[19]之前,一定要进行全面考虑,包括工作量、成本、时间和仪器设备的选择与维护。
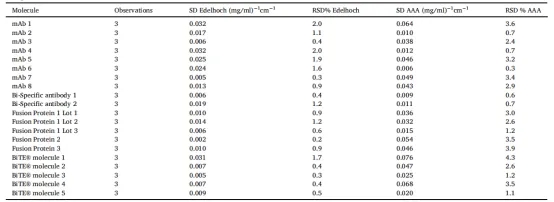
表3 Edelhoch法和AAA法消光系数SD和RSD的比较。
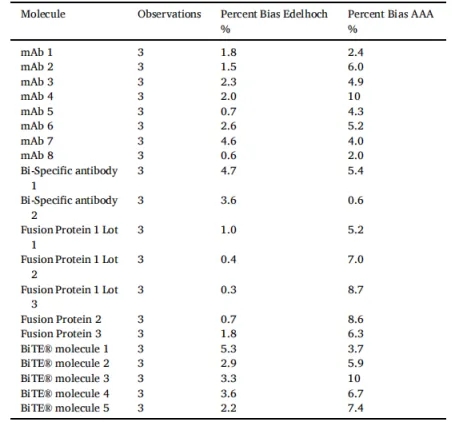
表4 Edelhoch法和AAA法的消光系数与理论消光系数的比较。
在这部分的研究中,数据显示Edelhoch方法的RSD值从0.2%到2.0%,平均RSD为1%,而AAA方法显示最低RSD为0.3%,最高RSD为4.3%,平均RSD为2.4%,如表3和图1所示。两种方法与理论消光系数的偏差如表4所示,用%偏差表示。在本研究中,Edelhoch法与理论消光系数之间,最大和最小偏差分别为5.3%和0.3%,平均偏差为2.4%(表4和图2)。AAA法与理论消光系数的最大偏差和最小偏差分别为10%和0.6%,平均偏差为6%。虽然AAA是一种有价值的方法,但它更大的局限性是该方法执行的复杂性,特别是在水解过程和随后的HPLC分析时。Edelhoch方法提供了一种方便的替代方案(在几个小时内完成),并显著提高了执行效率,降低了成本、时间,同时提高了安全性。
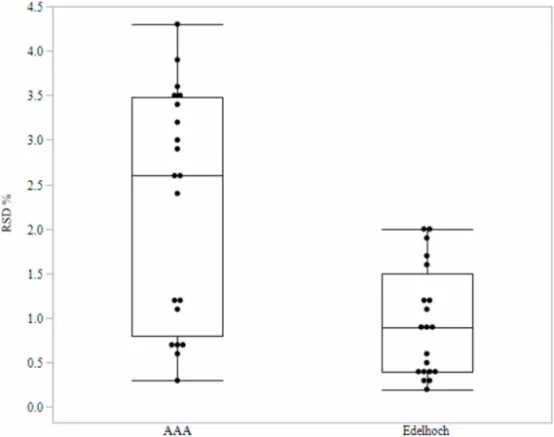
图1 比较AAA方法和Edelhoch方法的RSD%值的箱形图。每个点代表一个分子的RSD%。对于每种方法,方框显示了第25、50和75个百分位数。垂直线,或“须”,延伸到最小和最大的RSD%值(由表3生成)。
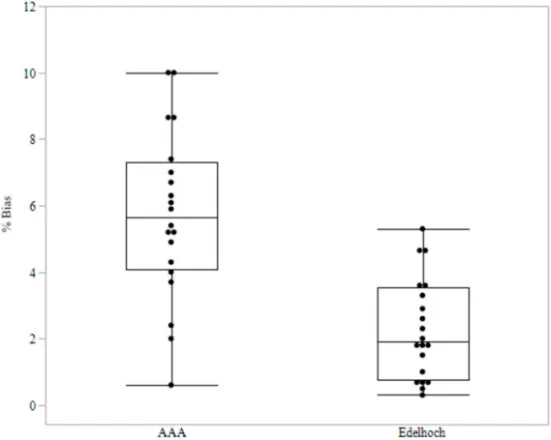
图2 比较AAA方法和Edelhoch方法的%偏差值的箱形图。每个点代表一个分子的偏差%。对于每种方法,方框显示了第25、50和75个百分位数。垂直线,或“须”,延伸到最小和最大的平均偏差值(由表4生成)。
结论
蛋白药物浓度的测定在药物开发过程中是至关重要的。目前有多种方法来确定浓度,最广泛使用的方法是采用在280 nm处的吸收。这反过来又需要可靠的方法来确定消光系数。蛋白药物的消光系数通常是在早期开发过程中通过实验来确定的。本研究采用Edelhoch方法确定了不同结构类别的大量蛋白药物的消光系数。数据结果表明,与AAA法相比,Edelhoch方法在测定蛋白质的实验消光系数方面,更有效、可靠、安全,同时降低了成本和时间。总的来说,Edelhoch方法是一种更容易获得和更可靠的方法,一般来说会更便宜、更容易实施,特别是对于初创公司的生物制药业务。
参考文献
[1] Beer A. Bestimmung der Absorption des rothen Lichts in farbigen Flüssigkeiten" [Determination of the absorption of red light in colored liquids]. Annalen der Physik und Chemie 1852; 86(5):78–88 (in German).
[2] Benson AM, Suruda AJ, Talalay PJ. Concentration dependent association of ketosteroid isomerase of Pseudomonas testosteroni. J Biol Chem 1975; 250:276–80.
[3] Jaenicke L. A rapid micro method for the determination of nitrogen and phosphate in biological material. Anal Biochem 1974;61(2):623–7.
[4] Nozaki Y. Determination of the concentration of protein by dry weight—a comparison with spectrophotometric methods 1986; 249:437–46.
[5] Edelhoch H. Spectroscopic determination of tryptophan and tyrosine in proteins. Biochemistry 1967;6(7):1948–54.
[6] Gill SC, Von Hippel PH. Calculation of protein extinction coefficients from amino acid sequence data. Anal Biochem 1989;182(2):319–26.
[7] Grimsley GR, Pace CN. Spectroscopic determination of protein concentration. Current protocols in Protein Science. John Wiley & Sons; 2003. p. 3.1.1–3.1.9.
[8] Wetlaufer DB. Ultraviolet spectra of proteins and amino acids. Adv Protein Chem 1962; (17):303–90.
[9] Scopes RK. Measurement of protein by spectrophotometry at 205 nm. Anal Biochem 1974; 59(1):277–82.
[10] Pace CN, Vajdos F, Fee L, Grimsley G, Grey T. How to measure and predict the molar absorption coefficient of a protein? Protein Sci 1995; 4:2411–23.
[11] Maity H, Wei A, Haider JN, Srivastava A. Comparison of predicted extinction coefficients of monoclonal antibodies with experimental values as measured by the Edelhoch Method. Int J biol molecules 2015; 77:260–5.
[12] Basale LY, Chennell P, Tokhadze N, Astier A, Sautou V. Physicochemical stability of monoclonal antibodies. J Pharmacol Sci 2020; 109:169–90.
[13] Evans AR, Capaldi MT, Goparaju G, Colter D, Shi FF, Aubert S, Li LC, Mo J, Lewis MJ, Ping Hu P, Alfonso P, Mehndirattaa P. Using Bispecific antibodies in forced degradation studies to analyze the structure function relationships of symmetrically and asymmetrically modified antibodies. mAbs 2019; 11:1101–12.
[14] Kontermann RE, Brinkmann U. Bispecific antibodies. Drug Discov Today 2015;20 (7):838–47.
[15] Levin D, Golding B, Strome SE, Sauna ZE. Fc fusion as platform technology; potential for modulating immunogenecity. Trends Biotechnol 2015; 33:27–34.
[16] Goebeler ME, Bargou RC. T cell-engaging therapies-BiTEs and beyond. Nat Rev Clin Oncol 2020; 17:418–34.
[17] Huels AM, Couplet TA, Sentman CL. Bispecific T cell engagers for cancer immunotherapy. Immunol Cell Biol 2015; 93930:290–6.
[18] Crabb JW, Ericsson L, Atherton D, Smith A, Kutny R. In: Villafranca JJ, editor. Current research in protein chemistry. New York: Academic Press; 1990. p. 49–61.
[19] Crab JW, West KA, Dodson WS, Hulmes JD. Current protocols in protein science. John Willey and Sons Inc); 1997. 11.9.1-11.911.9.42.
[20] Williams AP. General problems associated with the analysis of amino acids by automated ion exchange chromatography. J Chromatogr 1986; 373:175–90.
[21] Yuksel KU, Anderson TT, Apostol I, Fox JW, Raymond JP, Strydom DJ. The hydrolysis process and the quality of amino acid analysis. Tech in Prot Chem 1995; 6:185–92.
网 址:www.bioprocessanalytics.com
邮 箱:info@bioprocessanalytics.com



长按屏幕识别二维码
打开手机扫描二维码